
تالاسمی
انتقال اکسیژن در خون توسط گلبولهای قرمز و پروتئینی به نام هموگلوبین انجام می گیرد.
این پروتئین از دو زنجیره آلفا و دو زنجیره بتا تشکیل شده است.
این پروتئین از دو زنجیره آلفا و دو زنجیره ها که در اثر نقصان عملکرد ژن های تولید کننده آنها بروز می کند باعث بیماری کم خونی ژنتیکی موسوم به تالاسمی می شود.
تالاسمی بتا
تالاسمی بتا در اثر نقص در تولید زنجیره بتا ایجاد می شود.
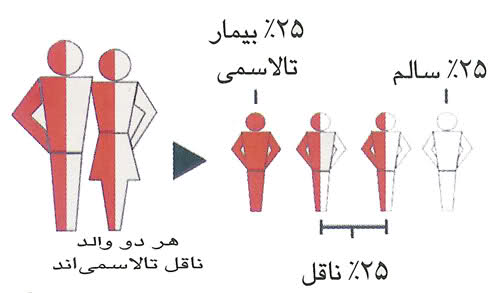
وراثت این بیماری به صورت اتوزومال مغلوب است و در نتیجه والدین ناقل به احتمال 25 درصد ممکن است صاحب فرزند بیمار شوند.
تشخیص پیش از تولد تالاسمی بتا در دو مرحله تشخیص ناقلین (مرحله اول) و تشخیص پیش از تولد( مرحله دوم) انجام می گیرد.
انواع تالاسمی بتا
تالاسمی مینور
در این نوع تالاسمی کمبود پروتئین به حدی نیست که عملکرد هموگلوبین ها را تحت تاثیر قرار دهد. فردی که به این بیماری دچار شده است گویی فقط کم خونی خفیف را تجربه می کند.
تالاسمی بینابینی
در این نوع تالاسمی فرد دچار کم خونی نسبتا شدید می شود و علائمی مثل ،بزرگی طحال و بدفرمی استخوان ها می تواند از نشانه های این نوع تالاسمی باشد. چون در این نوع تالاسمی فرد نیاز به تزریق خون به طور دوره ای دارد ،می توان تالاسمی بینابینی را در گروه تالاسمی ماژور قرار دهیم. تزریق خون برای بیماران تالاسمی به بهبود کیفیت زندگی بیماران کمک می کند وکمکی برای از بین بردن این بیماری نمی کند .
تالاسمی ماژور
تالاسمی ماژور شدیدترین نوع تالاسمی بتا می باشد که گاهی منجر به مرگ بیمار می شود.این نوع تالاسمی در طفولیت شروع می شود و علاوه بر انتقال خون به طور منظم ،نیاز به مراقبت های طبی فراوانی دارد.
فرد مبتلا به تالاسمی ماژور یک ژن بیماری را از پدر و ژن دیگر بیماری را از مادر به ارث برده است.این بیماری معمولاً در 6 ماه اول زندگی کودک نمایان می شود و به علت خون سازی غیر موثر،کبد و طحال شروع به خون سازی می کنند و بزرگ می شوند. در صورت عدم شروع تزریق خون، بافت های مغز استخوان و مکان های خون ساز خارج از مغز استخوان فعال و بزرگ شده و موجب بزرگی مغز استخوان ها به خصوص استخوان های پهن و بزرگی کبد و طحال می شود.
علائم تالاسمی ماژور
معمولا این علائم از ۶ ماهگی به بعد ظاهر می شود که شامل رنگ پریدگی، بی حالی و ضعف کودک است ،که با بزگ تر شدن کودک این علائم شدیدتر می شوند. پهن شدن استخوان چهره ،تغییر چهره بیمار، بزگ شدن کبد و اختلالات رشد از نشانه های تالاسمی ماژور هستند.
این کودکان بخاطر کم خونی ،به صورت مداوم نیاز به تزریق خون دارند و چون تزریق مداوم خون باعث افزایش آهن بدن می شود لذا لازم است ماده دفع کننده آهن بنام آمپول دسفرال نیز برای این کودکان تزریق شود.
درمان تالاسمی ماژور
انتقال خون
انتقال به طور دوره ای و منظم برای درمان تمام اشکال تالاسمی بسیار موثر است.با کمک این تزریق های منظم خون ،مقادیر زیادی از سلول های قرمز خونی سالم و هموگلوبین طبیعی که قادر به انتقال اکسیژن هستند تولید می شوند.
آهن راه طبیعی برای حذف در بدن ندارد ، بنابراین آهن موجود در خون تزریق شده، در بدن انباشته می شوند و وضعیتی را به نام افزایش غیر طبیعی آهن در بدن ایجاد می کنند.
آهن بیش از حد در بدن برای ارگان های بدن مثل قلب و کبد سمی است و می تواند منجر به مرگ زودرس و یا نارسایی ارگان ها در فرد بیمار شود.افزایش غیر طبیعی آهن ، به ویژه در قلب مهمترین علت مرگ و میر بیماران تالاسمی ماژور است.
Chelation Therapy
برای دفع آهن اضافی که از طریق تزریق به بدن بیمار وارد می شود ،بیمار تخت درمان سخت و دردناک با دارویی به نام دسفرال قرار می گیرد.این دارو از طریق سوزن متصل به پمپ و زیر پوست ناحیه معده و یا پاها تزریق می شود.
این روش درمان در هفته به مدت ۵ یا ۷ روز و به مدت ۱۲ساعت طول می کشد.
تالاسمى آلفا
تالاسمی آلفا به علت کاهش میزان زنجیره آلفاگلوبین ایجاد می شود.این بیماری به صورت اتوزوم مغلوب به ارث می رسد.
تشخیص ژنتیکی این بیماری در دو مرحله تشخیص ناقلین(مرحله اول) و تشخیص پیش از تولد(مرحله دوم) انجام می گیرد.
افراد سالم 4 نسخه ژن فعال آلفا ((دوتابر روی هرکدام از کروموزوم 16) دارند.
تالاسمی آلفا درواقع زمانی اتفاق می افتد که یکی از چهار ژن لازم برای ساخت زنجیره ی گلوبین آلفا وجود نداشته باشد و یا نرمال نباشد.تالاسمی آلفا شدیدترین نوع تالاسمی است که این نوع تالاسمی ممکن است باعث سقط جنین شود.
تالاسمی کلا دو نوع اصلی دارد،تالاسمی آلفا و تالاسمی بتا که هر کدام از آنها دو نوع وخیم و خفیف دارند،که نوع خفیف تالاسمی، تالاسمی آلفا مینور نیز نامیده می شود .نوع وخیم تالاسمی هم معمولا در اوایل کودکی تشخیص داده می شود و برای تمام عمر در فرد باقی می ماند.ژن های تالاسمی از پدر و مادر به فرزندان منتقل می شوند،از هر والد دو ژن دریافت می شود و تالاسمی آلفا زمانی اتفاق می افتد که یک یا دوتا از این ژن ها وجود نداشته باشد.در افراد مبتلا به تالاسمی ژن هایی که هموگلوبین تولید می کنند با ژن های معمولی متفاوت هستند.
.jpg)
انواع تالاسمی آلفا
1-فردی که یک نسخه ژن غیر فعال و 3 نسخه ژنی فعال داشته باشد، به عنوان ناقل خاموش آلفا تالاسمی شناخته می شود.
2-افرادی که دو نسخه فعال و دو نسخه غیر فعال داشته باشند، ناقلان دارای خصیصه آلفا تالاسمی نامیده می شود. این افراد دچار کم خونی خفیف هستند که نشانه بالینی ندارد و معمولاً با فقر آهن اشتباه گرفته می شود.
3-دو نوع مهم آلفا تالاسمی که با عوارض متعدد بالینی رو به رو هستند آلفا تالاسمی ماژور یا هیدروپس فتالیس و بیماری H است. مبتلایان به بیماری H در بدو تولد با علائمی چون کوچک بودن گلبولهای قرمز، کم خونی، همولیتیک هایپرکرومیک، بزرگی کبد و طحال، زردی و در مواردی تغییرات استخوانی مرتبط با تالاسمی رو به رو هستند. این بیماری نیاز به تزریق خون دارند.در جنین های مبتلا به هیدروپس فتالیس یا تالاسمی آلفا ماژور معمولاً پیش از تولد یا مدت کوتاهی پس از تولد فوت می کنند.
بررسی ژنتیکی : در ازدواج های فامیلی و در افرادی که تشخیص تالاسمی مینور دارند در هنگام ازدواج نیاز به
بررسی ژنتیکی ژن های بتا و آلفا تالاسمی دارند.
همچنین در خانواده هایی که یک فرزند مبتلا به تالاسمی دارند.
تهیه کننده : دکتر زهرا پاکزاد -فائزه سرور